
 Polycystic kidney disease is a genetic abnormality that causes multiple cysts to form on the kidneys and sometimes in other parts of the body. The disease has two classifications depending on the specifics of the transferability of the defective genes. Autosomal dominant polycystic kidney disease (ADPKD) is by far the more common form of the disorder, afflicting an estimated one to two in 1,000 live births. Autosomal dominance in a hereditary characteristic means that only one parent must have the gene in order to pass it on to offspring. The other form of polycystic kidney disease is autosomal recessive PKD (ARPKD). An autosomal recessive trait is one that requires genetic donations from both parents. ARPKD is found in only about 1 in 20,000 live births.
Polycystic kidney disease is a genetic abnormality that causes multiple cysts to form on the kidneys and sometimes in other parts of the body. The disease has two classifications depending on the specifics of the transferability of the defective genes. Autosomal dominant polycystic kidney disease (ADPKD) is by far the more common form of the disorder, afflicting an estimated one to two in 1,000 live births. Autosomal dominance in a hereditary characteristic means that only one parent must have the gene in order to pass it on to offspring. The other form of polycystic kidney disease is autosomal recessive PKD (ARPKD). An autosomal recessive trait is one that requires genetic donations from both parents. ARPKD is found in only about 1 in 20,000 live births.
Symptoms
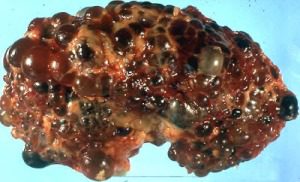
Most of the time (although not every time), ADPKD symptoms manifest in utero or very shortly after birth. The disorder results in cysts forming on the kidneys. Cysts are fluid-
renal failure requiring dialysis.
Cysts can also form on other organs as a result of ADPKD, including the liver, pancreas, spleen, brain, ovaries, prostate, cardiac valve, colon, and aorta. The symptoms of advanced ADPKD involve failure of function of any of these organs, most commonly the kidneys. These complications are potentially lethal, causing death by toxicity from failure of renal or hepatic function, or from a heart attack. Other complications include high blood pressure and increased frequency of urinary tract infections.
Diagnosis
Diagnosis of ADPKD relies on family history, medical imaging, and genetic testing. Because of the autosomal dominant nature of the genetic disorder, it is almost always found in families; a dominant genetic trait cannot be a “sleeper” gene carried by a parent who does not manifest symptoms. In the case of ADPKD the picture is somewhat complicated by the fact that two different genetic anomalies are responsible for
the condition, but more than 90 percent of patients with ADPKD have family members also exhibiting the trait. A history of ADPKD in the family is a risk factor calling for monitoring.
Medical imaging can reveal enlargement and the formation of cysts on the kidneys, both of which are primary symptoms of ADPKD. A final confirmative diagnosis can be provided by a genetic assay to reveal the presence of the genetic causes of the disorder.
Treatment
There is no cure for PKD, but treatments can relieve symptoms and extend life expectancy. Surgical treatments can reduce or drain the cysts and relieve pain. Pain medications can also help relieve the discomfort. Aggressive efforts to lower blood pressure can reduce or slow down progression towards end-
When the disease has progressed to renal failure, it is treated as with any other form of kidney failure, by dialysis or by a kidney transplant. Kidney transplant is a standard treatment for final-